Phylogenetics: BayesTraits Lab

|
EEB 349: Phylogenetics |
| In this lab you will learn how to use the program BayesTraits, written by Andrew Meade and Mark Pagel. BayesTraits can perform several analyses related to evaluating evolutionary correlation in discrete morphological traits. This program is meant to replace the older programs Discrete and Multistate. You will learn not only how to use the program on the Windows-based PCs in the computer lab, but also how to download and use it on the cluster (the cluster is better for long runs). |
We will use BayesTraits interactively for awhile on the PCs in the computer room (Part 1), then we will set up a non-interactive run on the cluster in Part 2 so that you know how to do this.
Contents
Part 1: Running BayesTraits on your own laptop
Download BayesTraits
Download BayesTraits from Mark Pagel's web site, click on the "Software" link, then click on the "Description and Downloads" link under "BayesTraits". Download the version specific to your platform. BayesTraits will unpack itself to a folder containing the program itself along with several tree and data files (e.g. Primates.txt and Primates.trees). I will hereafter refer to the folder containing these files as simply the BayesTraits folder. Go back to Mark Pagel's web site and download the manual for BayesTraits. This is a PDF file and should open in your browser window.
Download the tree and data files
For this exercise, you will use data and trees used in the SIMMAP analyses presented in this paper (you should recognize the names of at least two of the authors of this paper):
Jones C.S., Bakker F.T., Schlichting C.D., Nicotra A.B. 2009. Leaf shape evolution in the South African genus Pelargonium L'Her. (Geraniaceae). Evolution. 63:479–497.
The data and trees were not made available in the online supplementary materials for this paper, but I have obtained permission to use them for this laboratory exercise. The links below are password-protected, so ask us for the username and password before clicking on the links:
You should move these files to the aforementioned BayesTraits folder so that they can be easily found by the BayesTraits program.
Assessing the strength of association between two binary characters
The first thing we will do is see if the two characters (leaf venation and leaf dissection) in pelly.txt are evolutionarily correlated. The leaf venation trait comprises two states: 0 means leaves are palmately veined (several major veins meet at the base of the leaf), and 1 means leaves are pinnately veined (one main vein runs down the long axis of the leaf blade). The leaf dissection trait also has two states (I've merged some states in the original data matrix to produce just 2 states): 0 means leaves are unlobed or shallowly lobed, and 1 means leaves are lobed, deeply lobed, or dissected.
To test whether these two traits are correlated, we will estimate the marginal likelihood under two models. The independence model assumes that the two traits are uncorrelated. The dependence model allows the two traits to be correlated in their evolution. The model with the higher marginal likelihood will be the preferred model. You will recall that we discussed both of these models in lecture, and also discussed the harmonic mean method that BayesTraits uses to evaluate models (you may wish to pull up those lectures to help answer the questions that you will encounter momentarily).
If you are using Windows, start BayesTraits by opening a console window , navigate to the BayesTraits directory, and type the following to start the program:
BayesTraits pelly.tre pelly.txt
If you are using a Mac, start BayesTraits by opening a terminal window, navigate to the BayesTraits directory, and type the following to start the program:
./BayesTraits pelly.tre pelly.txt
You should see this selection appear:
Please Select the model of evolution to use. 1) MultiState 2) Discrete: Independent 3) Discrete: Dependant 4) Continuous: Random Walk (Model A) 5) Continuous: Directional (Model B) 6) Continuous: Regression 7) Independent contrast
Press the 2 key to select the Independent model. Now you should see these choices appear:
Please Select the analysis method to use. 1) Maximum Likelihood. 2) MCMC
Press the 1 key to select maximum likelihood. Now you should see some output showing the choices you explicitly (or implicitly) made:
Options:
Model: Discete Independant
Tree File Name: pelly.tre
Data File Name: pelly.txt
Log File Name: pelly.txt.log.txt
Summary: False
Seed 3162959925
Analsis Type: Maximum Likelihood
ML attempt per tree: 10
Precision: 64 bits
Cores: 1
No of Rates: 4
Base frequency (PI's) None
Character Symbols: 00,01,10,11
Using a covarion model: False
Restrictions:
alpha1 None
beta1 None
alpha2 None
beta2 None
Tree Information
Trees: 99
Taxa: 154
Sites: 1
States: 4
Now type run to perform the analysis, which will consist of estimating the parameters of the independent model on each of the 99 trees contained in the pelly.tre file.
- On p. 11, where does Pagel get his rule of thumb that says "...if this [constrained] model is two or more log-likelihood units worse than the unconstrained model, then the two rate coeficients differ."? (hint: what would the likelihood ratio test statistic be in this case, how many degrees of freedom are there, and what would the significance probability be in this case?)
- In the section Reconstruct an ancestral state under Using maximum likelihood: Given that we are using an unrooted tree, would you agree or disagree with the statement "The attraction of MRCA’s is that one exists in each tree"?
- After conducting a reversible-jump analysis, the results are stored in the file MatingSystem.txt.log.txt. One of the columns is labeled "model" and has values that are either '00 or '0Z. If you wanted to know the marginal posterior probability of the model 00 (corresponding to
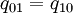 ), how would you go about figuring this out from the output provided?
), how would you go about figuring this out from the output provided? - At the very end of the section Reconstruct an ancestral state under Using Markov chain Monte Carlo: the manual says "Note: Harmonic means can be unstable so this analysis should be repeated maybe 5 times and with very long runs (10s or 100s of million of iterations) to be sure of the result." Assuming you get a very stable harmonic mean value over 5 runs, as recommended. Does this mean that you will accurately assess the Bayes factor?
Tutorial Notes
1. Remember throughout the tutorial to use Primates.first.tree instead of Primates.trees! Note that your output will only correspond to that of tree number 1 in the sample output from the BayesTraits manual.
2. BayesTraits must be run from the command line, which means you must open a command window to run the program. Simply double-clicking the BayesTraits executable file will cause it to run, but not for long! The problem is that when you double-click the program, you have no way to tell it what tree and data file to use, so it simply quits immediately. Open a Terminal window (Mac) or console window (Windows) in the directory where the BayesTraits executable is located. You can then start the program as follows:
BayesTraits Primates.first.tree MatingSystem.txt
Note however that later the tutorial switches to using the data fle Primates.txt, so at that point you should replace MatingSystem.txt with Primates.txt on the command line.
3. BayesTraits stops after it executes the run command, so you will need to restart it numerous times during the tutorial.
4. The output that you see scrolling by on the screen (especially during the MCMC analyses) is saved automatically in the file MatingSystem.txt.log.txt. This file is overwritten each time you run BayesTraits, so rename it before running BayesTraits again if you want to save it.
5. The default number of MCMC iterations is 5,050,000. This will take some time to run. For our purposes, it is ok to reduce this number. For example, to tell BayesTraits to only run for 550,000 iterations, type in the following command before you type run:
it 550000
6. There is a listing of all commands recognized by BayesTraits at the end of the manual.
Part 2: Running BayesTraits on the cluster
We will now switch to using the (older, Mac-based) cluster to run BayesTraits. BayesTraits is not installed on the cluster, so this provides a good opportunity to show you how to download and unpack software into your home directory and run programs not already installed for you.
BayesTraits is different than most of the programs used on the cluster in that it is interactive: it expects you to type some commands after it starts. This is difficult when you use qsub to submit a job to the cluster since you are not there to supply these commands once it starts. This part of the lab shows you how to get around this difficulty when using programs like BayesTraits.
Downloading and unpacking BayesTraits
First, connect to bbcxsrv1.biotech.uconn.edu to get a command prompt (using either putty.exe on Windows or the ssh command in your Mac Terminal application). The full URL to the OS X PPC version of BayesTraits is
http://www.evolution.reading.ac.uk/Files/BayesTraits-OSX-PPC-V1.0.tar.gz
If you had a browser open, and you typed in this URL, your browser would save the file BayesTraits-OSX-PPC-V1.0.tar.gz on your hard drive. But you are not using a browser on the cluster, you are logged in using secure shell.
You could download the file to your PC, then upload it to the cluster using FileZilla (Windows) or Fugu (Mac), but let's instead use the curl command:
curl http://www.evolution.reading.ac.uk/Files/BayesTraitsV2-Beta-Linux64.tar.gz > BayesTraitsV2-Beta-Linux64.tar.gz
This tells curl to access the specified URL (curl will stand in for a web browser) and the -o option says to save the resulting file as BayesTraits-OSX-PPC-V1.0.tar.gz.
Once you have the file in your home directory (use the ls command to check), you will need to unpack it using the tar command:
tar zxvf BayesTraits-OSX-PPC-V1.0.tar.gz
The file extension .tar.gz is very common for software targeting Linux and Mac OS X systems. This extension has a very specific meaning: the .tar part means that the file is actually an archive comprising several files saved one after the other (tar stands for "tape archive", even though no one uses magnetic tapes anymore). The .gz part means that the archive has been compressed using the gzip program. The command line switches for tar (zxvf) are decoded as follows:
- z means unZip the archive first
- x means eXtract the component files
- v means be Verbose and tell me what's going on as you work
- f simply means that the name of the File to extract follows immediately afterwards
Once the tar command has completed, you should have a directory named BayesTraits. Use the cd command to move into that directory, then use the ls command to see what's there. You should see the same 5 files as before.
Running BayesTraits
We will do a very simple analysis just so you will know how to run BayesTraits in batch mode. Because analyses on the cluster are submitted via the qsub command, and thus no one will be present to answer those questions BayesTraits asks when it runs, we'll supply the answers in a file (arbitrarily named commands.txt) and feed that file to BayesTraits when it is invoked.
Create the commands.txt file
Create a file inside the BayesTraits directory using the pico editor named commands.txt, and save the following in the file:
# choose the model (1 = multistate, 2 = independence, 3 = dependence) 3 # choose the method (1 = ML, 2 = MCMC) 2 ratedev 10 rjhp exp 0 30 run
The lines starting with a pound sign (or hash) are comments and are optional. Otherwise, this file just presents to BayesTraits exactly what you would type if you were running the program interactively.
Create the qsub script
As you know by now, you use the qsub command to submit a job to the cluster. The advantage of using the qsub command is that it will start your job on a processor that is currently idle (assuming there are idle processors available in the cluster). Not using qsub results in your job being started on the head node of the cluster. The head node is the processor that everyone uses to interact with the cluster, and if you start a run there, it has a fair chance of simply being killed as soon as the system administrator (Jeff Lary) notices it because it makes the head node appear very sluggish to other users!
The qsub command accepts the name of a script file that carries out your wishes, so we must first create this script (use pico to create and save the following in your home directory as a file named btgo, for example):
#$ -o junk.txt -j y #$ -m bea #$ -M your.name@uconn.edu cd $HOME/BayesTraits ./BayesTraits Primates.trees Primates.txt < commands.txt
You have experienced these scripts before. The first few lines begin with the sequence of characters #$, which alerts qsub that a qsub command is coming. The first command is the -o junk.txt -j y part, which tells qsub that you want the output of the run saved in a file named junk.txt and you would like any error message appended to this same file (the -j y part is indeed cryptic, but stands for "join [standard error to output] yes"). Be sure to delete or rename any file named junk.txt that already exists.
The second line says to send an email notification before, at the end of, and in case of abort.
The third line tells the Sun Grid Engine where to send the email notifications. You should of course change this to your own UConn email address Email addressed outside UConn will be ignored.
The fourth line says to cd into the BayesTraits directory (this means that the BayesTraits folder will now be the working directory, and any output files created by the program will be saved there).
The fifth line actually starts the program. Since we are running this on the cluster and are not so limited by time, we'll use the full Primates.trees file (containing 500 trees). Note the very important commands.txt part, which feeds the answers to all questions to the BayesTraits executable. Also, note the period-slash (./) before the name of the program. This says that the program can be found in the current directory (the current directory is signified by a dot; one of the crazy things about unix is that the current directory is not searched by default when looking for files!).
Submit the script
Now submit the job as follows:
qsub btgo
As before, you can periodically check to see if it is still running using the qstat command. You can use pico to look at the junk.txt file in your home directory, or look in the BayesTraits directory and examine the files created there as the program runs. A useful command is tail:
cd $HOME/BayesTraits tail Primates.txt.log.txt
This shows you only the last few lines of the file listed. To show the last 100 lines, you can use the -n switch:
tail -n 100 Primates.txt.log.txt